What do we treat

Congenital anomalies of the esophagus and trachea.
There are several kinds:
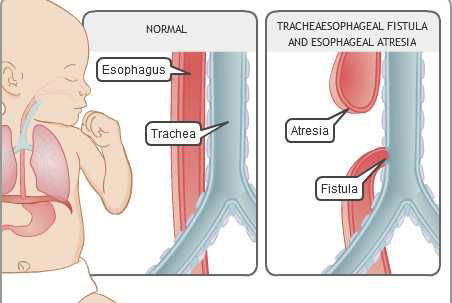
- Esophageal atresia- With esophageal atresia, esophagus does not form properly, resulting in two segments that don't connect to each other. Since the esophagus is in two segments, liquid that a baby swallows cannot pass normally through the esophagus and reach the stomach. Also, ingested liquids may spill over into the bronchial tubes and cause pneumonia or airway obstruction.
- Tracheoesophageal fistula (TEF)- Often occurs with esophageal atresia. It's an abnormal connection in one or more places between your child's esophagus and the trachea. Normally, the esophagus and the trachea are two separate tubes that are not connected, but if a TEF is present, stomach content may regurgitate into the lungs. It is also possible, though rare, that a TEF can be present from the upper end of the esophagus to the trachea in which event, swallowed liquids would pass directly into the lungs.
- Isolated tracheoesophageal fistula- (Very rare) Where there is an abnormal communication between an otherwise intact esophagus and trachea either as a narrow opening or there can also be a rare long communication between esophagus and trachea called a tracheoesophageal cleft.
- Trachea atresia- (Very rare) an absence of part or all of your child's trachea.
- Tracheal stenosis- (Very rare) is a constriction of the trachea.
- Tracheal occlusion- (Very rare) is a blockage in the trachea.
Intestinal atresias
Intestinal atresias are malformations of the intestines in which a segment of bowel is very narrow or is disconnected from the rest of the GI tract. Most commonly, these occur in or near the duodenum, just below the stomach. Although atresias are rare, babies born with this condition are often small for their gestational age and some may also have Down syndrome. Depending on exactly where the malformation is, bile may be released into the GI tract with nowhere to go but up, appearing in the esophagus. As with esophageal atresia, the malformation is repaired with surgery, though the exact nature of the malformation may mean surgery will be performed sooner rather than later.
Malrotation with volvulus
Until about the 10th week of pregnancy, a fetus’s GI tract develops, in part, in the umbilical cord. At this point, it returns to the abdomen and rotates 90 degrees to the right. The individual components of the GI tract, including the duodenum, which connects the stomach and the intestines, and the intestines themselves, rearrange themselves and begin to settle into a position that will then remain unchanged for the rest of a person’s life. At the end of this process, the GI tract is normally fixed; that is, it is contained and supported, and does not move.
Occasionally, this series of maneuvers and migrations is not performed properly and part of the GI tract, though still connected, ends up in the wrong place. This malrotation, as it is called, can also leave the GI tract unfixed. In some cases, this is not a problem; some people end up leading a normal life with an unfixed and rotated bowel. However, many babies have severe symptoms. Occasionally, as the GI tract settles itself into place, it loops around itself, reducing its blood supply or causing an obstruction. This is called volvulus. Malrotation with volvulus requires emergency surgery to correct the problem.
Hirschsprung’s disease
Hirschsprung’s disease is a condition in which nerve cells called ganglia have not formed on the inner wall of the bowel. This causes the bowel to contract and not relax, obstructing the lower intestine. Boys are about 10 times more likely to have the disease than girls. Again, surgery is used to correct the malformation. Surgeons will identify the bowel section without ganglia, cut it out, and reattach the two ends of healthy bowel. Sometimes a colostomy, or the surgical removal of some of the bowels, is necessary, and the surgeons will do the final repair at six to 12 months of age. Many babies who have undergone this procedure will develop and lead normal lives, since enough functioning bowel remains for digestion. However, a small number of babies with Hirschsprung’s disease also have an inflammation of the large intestine called colitis, which may complicate surgery and can be life threatening.
Anorectal malformations
Some babies are born with malformations of the anus, rectum, or both. There are several general types. They have varying degrees of severity and are treated with surgery. These malformations are often specific to boys and girls. The malformation can come in the form of an absence of an opening where the anus should be; a fistula, or small opening from the rectum to the urinary tract or to the vagina; and many variations from these general categories. Depending on the exact nature and severity of the malformation, babies may be left completely continent, with full control over their bowel movements; partially continent; or incontinent. In general, surgeons will close the fistula and, in the case of a missing anus, create an opening and gently pull though the bottom of the bowel, creating a new anus.
Abdominal wall defects
A defect in the abdominal wall may allow some of the digestive system to develop outside the baby’s body in the amniotic fluid of the womb. These rare abnormalities are usually very small and only a small portion of the intestines are exposed. Gastroschisis occurs on the abdominal wall. Omphalocele occurs on the umbilicus, or belly button. Each of these defects allows some of the digestive system to develop outside the body. They can be small or large, and involve one organ or several.
Sometimes, these conditions can be diagnosed before birth. Exposure to the amniotic fluid can cause damage to the intestines. For this reason, some hospitals may suggest a planned early caesarean section to limit the extent of intestinal exposure to amniotic fluid. There is still debate about whether this is the best course of action. The treatment is surgical reinsertion of the intestines into the body. Depending on the size and extent of the condition, there may be more than one surgery needed to accomplish this goal.
Congenital biliary anomalies
Although considered congenital anomalies, biliary atresia and choledochal cyst may both be acquired conditions occurring or developing within the first few weeks of life.
Biliary atresia
Biliary atresia presents in the first few weeks of life. It is thought that an inflammatory process from an unknown cause affects the bile duct in the newborn infant. There is variable destruction of the extrahepatic bile ducts, causing obstructive jaundice and liver failure. Surgical correction of this abnormality before 8 weeks of age produces the best outcome.
Other anomalies associated with biliary atresia include intestinal malrotation and genitourinary anomalies. An aberrant communication between the biliary and pancreatic ducts has been reported in association with the condition, but it is not clear whether this is aetiologically important. Correction of this condition is by hepaticojejunostomy when a segment of extrahepatic duct is patent, or by porto-enterostomy for the more common severe case. In the latter operation, a jejunal Roux-en-Y loop is anastomosed to the porta hepatis after excision of the atretic duct remnants in the porta. Liver transplantation is frequently required for severe cases and atresia is the commonest indication for paediatric transplantation. Excellent long-term results are achieved with transplantation.
Choledochal cyst
Cystic dilatation of the intra- or extrahepatic ducts is a rare condition, usually presenting before the age of 16 years. In 20% of cases, presentation is in adult life. Symptoms produced by the cyst include cholangitis, pancreatitis, stone formation and jaundice. Infants may occasionally present with an abdominal mass. The cause of this condition is debated. Anomalous communication between the biliary and pancreatic ducts is frequently found, but a degree of distal biliary obstruction may also be aetilogically important in the development of the cyst.
Cysts are classified according to their site and shape, although 80% are fusiform abnormalities of the extrahepatic bile duct (Classification of choledochal cysts (after Todani, Alonso-Lej).). Type II cysts are extremely rare.
The preferred surgical method of treatment is complete cyst excision with biliary reconstruction, because of the risk of malignant transformation if the cyst is left in situ. The rate of malignant transformation may be as high as 25% for untreated cysts. When multiple intrahepatic cysts are present complete excision may not be possible and treatment consists of producing good biliary drainage by excising any extrahepatic component.
Congenital Chest Wall Deformities
There are two basic types of congenital chest wall deformities: Pectus Excavatum (sunken chest), and Pectus Carinatum (a protuberant or "pigeon chest").
Some patients are born with a combination of the two, which creates an asymmetric deformity. While both kinds of patients may have chest pain from the deformity, excavatum patients commonly complain of shortness of breath and early fatigue during aerobic exercise.
Pectus Excavatum (Sunken Chest)
Traditionally, large incisions have been made across the front of the chest to allow the resection of abnormally shaped rib cartilage and a surgical cut (osteotomy) across the sternum (breastbone). This procedure allowed the deformity to be corrected but resulted in scarring and long term rigidity of the chest wall. Surgeons in the Division of Pediatric Surgery are utilizing newer, minimally invasive approaches to correct congenital chest wall deformities.
Pectus Excavatum is now repaired utilizing the Nuss approach, which works like braces on misaligned teeth to lift the sternum and reshape the chest wall. The U-shaped bar is introduced from the side of the chest, using thoracoscopic visualization for optimal placement. The bar is left in place for two years and is then removed as an outpatient procedure.
In 1999, Dr. Nuss, who invented the technique, traveled from his hospital in Virginia to visit Children's Hospital Los Angeles. Our pediatric surgeons took advantage of this visit and, for the past 10 years, have been using this procedure to correct Pectus Excavatum. Since then, the technique has been used in nearly 100 patients.
Ideally, the Nuss procedure is performed between 10 and 14 years of age, before the chest wall becomes rigid. Postoperatively, the patient’s ability to breathe and exercise is often significantly improved within a few months.
In the future, it is possible that excavatum defects may be corrected utilizing an implantable magnet which will allow for the correction of excavatum defects at an even earlier age. This research is ongoing and initial results are encouraging.
Pectus Carinatum (Pigeon Chest)
Pectus Carinatum can now be corrected using an externally applied, custom made brace. Pediatric surgeons at our hospital have teamed with a local orthotics company to create a compression brace, which allows protuberant chest wall deformities to be reduced without surgery. The greatest success occurs in patients who are less than 15 years of age, ideally less than 12 years of age, when the chest walls are more flexible. Patients who wear their brace 23 hours per day typically see improvements in the first month. The brace is continually adjusted until the chest wall is flat, which typically occurs in three to six months.
Lung Malformations
Lung malformations, including congenital pulmonary airway malformations (CPAM, formerly called CCAM) and bronchopulmonary sequestration (BPS), are uncommon disorders that can cause a wide range of problems, including breathing difficulties, recurrent infection, and more rarely, cancer.
What is a lung malformation?
A lung malformation is a mass of disorganized lung tissue that forms as the lungs of the fetus develop in the womb. The mass, or lesion, usually involves only one of the lobes of the lung. There is no known cause.
In the past, these malformations have been classified into two main types. One is called a congenital cystic adenomatoid malformation or CCAM. More recently, CCAMs are also referred to by another name, CPAM, which stands for congenital pulmonary airway malformation. These lesions can consist of many tiny cysts (microcystic), or fewer large cysts (macrocystic), or a combination of both. Although most of these lesions stop growing around the 26th week of gestation, some continue to grow so patients’ need to be monitored closely.
The other type of lung lesion is called a bronchopulmonary sequestration, or BPS. This type of mass has an abnormal blood vessel that enters the mass directly from the aorta. Sometimes, there is too much blood flow to this abnormal lung and this may cause fetal heart failure.
There are other lung malformations with some differences. These may be a combination of the CPAM or BPS, called a hybrid lesion. There are also bronchogenic cysts, bronchial atresia, and congenital lobar emphysema. No matter the type, most are evaluated and managed in a similar way during fetal life.
Congenital anomalies of urinary tract
The kidney
The kidney is the most common site of congenital abnormalities. Some cause no problems but many result in impaired renal function. Renal malformations are often associated with other congenital defects - eg, a grossly deformed pinna with ipsilateral abnormalities of the facial bones.
- Renal agenesis:
- Virtually always unilateral.
- Kidney is either absent or undeveloped.
- It usually causes no symptoms and is found incidentally.
- Renal hypoplasia:
- This may appear as one small kidney with the other one larger than normal.
- Small kidneys also have small arteries and are associated with hypertension requiring nephrectomy.
- Supernumerary kidneys:
- Third kidney is very rare and not to be confused with the relatively common unilateral duplication of the renal pelvis.
- Renal dysplasia and multicystic kidney:
- Multicystic kidney of the newborn is normally seen in only one kidney as an irregularly lobulated mass of cysts and usually absent or atretic ureter.
- Frequently associated with contralateral abnormalities, especially ureteropelvic junction obstruction.
- Dysplasia of the renal parenchyma is seen with urethral obstruction or reflux present early in pregnancy, or obstructed ureter.
- Adult polycystic kidney disease.
- Infantile polycystic kidneys.
- Simple (solitary) renal cyst:
- This may be inherited or acquired.
- Significant renal damage is rare and usually only requires continuous follow-up.
- Renal fusion:
- Prevalence is 1 in 1,000 people.
- The most frequent abnormality seen is a horseshoe kidney containing two excretory systems and two ureters.
- They are usually asymptomatic but are prone to obstruction.
- Ectopic kidney:
- In simple ectopy, the kidney does not ascend properly and is found in the pelvis or over the brim.
- It is prone to obstruction and infection.
- Less common is crossed ectopy without fusion. The kidney then lies on the opposite side and is not attached to the normally placed kidney.
- Abnormal rotation: this rarely leads to renal disease.
- Medullary sponge kidney.
- Potter's syndrome.
Abnormalities of the ureter and ureteropelvic junction
These are relatively common and range from complete absence to duplication of the ureter.
- Ureteral atresia:
- The ureter may be absent or fail to extend to the bladder (therefore with a blind ending).
- It is associated with ipsilateral absent or multicystic kidney.
- Bilateral atresia is incompatible with life. Unilateral atresia is usually asymptomatic but may cause hypertension.
- Duplication of the ureter:
- This is one of the most common congenital malformations of the urinary tract, with duplication found in 0.9% of a series of autopsies.
- It is more common in females and is often bilateral.
- It is often asymptomatic but commonly presents with persistent or recurrent urinary tract infections.
- Ureterocele:
- This is a sacculation of the bladder end of the ureter that can occur either in the bladder or ectopically.
- It is much more common in girls than in boys.
- In 10% of cases it is bilateral.
- It may be asymptomatic or cause obstruction or incontinence or infection.
- Ectopic urethral orifice:
- This usually occurs with ureterocele and duplication of the ureter but single ectopic ureters are seen.
- Boys may present with epididymitis, as the ureter drains directly into the vas deferens or seminal vesicle.
- Girls normally present with incontinence with continual dribbling despite normal voiding.
- Obstruction of the ureteropelvic junction:
- This is a common congenital abnormality of the ureter, seen more often in boys than in girls. Most cases are unilateral and usually on the left.
- It is nearly always caused by a kink in the ureter where it meets the dilated renal pelvis.
- It can be diagnosed in utero.
- In children, it normally presents with pain and vomiting.
- Obstructed mega-ureter:
- This is caused by obstruction at the ureterovesical junction.
- It is four times more common in boys than in girls and is often bilateral.
- Often, it is associated with absent or dysplastic contralateral kidney.
- It is usually discovered during prenatal ultrasound.
- It frequently presents with haematuria, with symptoms of infection and abdominal pain.
Abnormalities of the bladder
- Bladder exstrophy:
- This is absence of the anterior wall of the bladder, with ureters delivering urine into the lower abdomen.
- It is associated with other abnormalities, especially epispadias.
- It requires surgical reconstruction.
- Persistent urachus:
- This may appear as a draining umbilical sinus and can become infected.
- Contracture of the bladder neck:
- This is a common cause of reflux, bladder diverticula and irritable bladder.
Anomalies of the penis and urethra in males
- Apenia:
- Congenital absence of the penis is very rare.
- More than 50% of patients have associated genitourinary anomalies - eg, cryptorchidism, renal agenesis and dysplasia.
- Megalopenis:
- The penis enlarges rapidly in childhood due to a high level of production of testosterone.
- Micropenis:
- This is a small but otherwise normally formed penis with a stretched length of less than 2.5 standard deviation below the mean. It is important to distinguish miscropenis from buried and webbed penis, which is usually of normal size. The most common aetiologies are:
- Hypogonadotropic hypogonadism: impaired secretion of gonadotrophin-releasing hormone (GnRH) by the hypothalamus occurs in some hypothalamic dysfunctions - eg, Kallmann's syndrome, Prader-Willi syndrome.
- Hypergonadotropic hypogonadism: the testes are functionally impaired - eg, gonadal dysgenesis.
- Idiopathic micropenis: there is normal hypothalamic-pituitary-testicular endocrine function.
- This is a small but otherwise normally formed penis with a stretched length of less than 2.5 standard deviation below the mean. It is important to distinguish miscropenis from buried and webbed penis, which is usually of normal size. The most common aetiologies are:
- Urethral stricture:
- This is uncommon; the two most common sites are the fossa navicularis and the membranous urethra.
- Severe cases can result in damage to the bladder and hydronephrosis due to back pressure of urine.
- Posterior urethral valves:
- A relatively frequent obstructive lesion in male neonates and infants.
- They are found at the distal prostatic urethra and mucosal folds, looking like thin membranes and causing varying degrees of obstruction when attempting to urinate.
- Boys usually present with a poor, intermittent, dribbling stream with frequent infection.
- Hypospadias:
- Urethral meatus is found on the underside of the penis.
- It occurs in around 1 in 300 male births.
- Around 7% of patients have an additional family member with hypospadias.
- Most cases occur on the distal penis or corona.
- Young children seldom report symptoms but older children and adults may complain of difficulty in directing the urinary stream, and of spraying.
- Around 10% of cases also have cryptorchidism.
- 9-15% of cases also have an open processus vaginalis or inguinal hernia.
- Epispadias:
- This consists of a defect of the dorsal wall of the urethra.
- The extent of the defect can vary from a mild glandular defect to complete defects, as are observed in bladder exstrophy and/or diastasis of the pubic bones.
- Epispadias occurs in 1 in 120,000 males and 1 in 450,000 females.
- The urethra is displaced dorsally so that it opens on to the top of the penis in males.
- Females have a bifid clitoris and separation of the labia.
- Incontinence is a common problem.
Female anomalies
- Distal urethral stenosis:
- This occurs in young girls with enuresis with slow and interrupted stream and recurrent infection.
- It is associated with secondary spasm of the external sphincter.
- Labial fusion:
- Fused labia minora can present with recurring urinary infection as a result of obstruction to the urine flow.
- Clitoral hypertrophy:
- This is caused by fetal exposure to androgens, usually caused by congenital deficiencies of adrenal enzymes of cortisol synthesis.
- Rarely, it may also be due to in utero exposure to progestational agents or idiopathic virilisation.
Anomalies of the testis
- Hypogonadism:
- Small testes with lack of development of secondary sexual characteristics, lack of libido and potency.
- Characteristically, patients are tall with long extremities.
- It is also associated with Klinefelter's syndrome.
- It is also associated with physiological problems and low intelligence.
- Ectopy and cryptorchidism:
- Ectopy: the testis has not followed the normal path of descent. By far the most common site is superficial inguinal.
- Cryptorchidism: testicular descent is arrested. At 1 year of age, nearly 1% of all full-term male infants have cryptorchidism, which is the most common congenital anomaly affecting the genitalia of newborn male infants. [1 ] The most useful classification of cryptorchidism is into palpable and non-palpable testes, and clinical management is decided by the location and presence of the testes:
- Retractile testes require only observation.
- Bilateral, non-palpable testes and any suggestion of sexual differentiation problems (eg, hypospadias) require urgent, mandatory endocrinological and genetic evaluation.
- Surgical orchidolysis and orchidopexy should be concluded at the age of 12 months, or 18 months at the latest.
- Spermatocele:
- This is a painless cystic mass found just above or posterior to the testis.
- They are mostly <1 cm in diameter and usually require no therapy.
- Varicocele:
- These are unusual in boys aged under 10 years and are more frequent at the beginning of puberty. They occur in around 14-20% of adolescents.
- They appear mostly on the left side (78-93% of cases).
- They are caused by dilatation of the pampiniform plexus (spermatic venous plexus).
- Fertility problems arise in around 20% of adolescents with a varicocele.
- Surgery is recommended for:
- Varicocele associated with a small testis.
- Additional testicular condition affecting fertility.
- Pathological sperm quality (in older adolescents).
- Bilateral palpable varicocele.
- Symptomatic varicocele.[1]
- Hydrocele:
- This is an excess of fluid between the two layers of the tunica vaginalis in young boys.
- It presents as a cystic mass which is often more tense at night.
- In the majority of infants, surgical treatment of hydrocele is not indicated within the first 12-24 months, as they usually resolve spontaneously.
- Early surgery is indicated if there is suspicion of a concomitant inguinal hernia or underlying testicular pathology.[1]
Chromosomal syndromes
- Turner syndrome.
- Other syndromes of gonadal dysgenesis:
- These range from bilateral streaks or bilateral dysgenetic testes to apparently normal testes.
- They may be asymmetrical - eg, streaked one side, normal the other. This is mixed gonadal dysgenesis.
Disorders of sex development
- The formerly called 'intersex disorders' have been changed to 'disorders of sex development' (DSD).
- This classification has arisen because of advances in knowledge of the molecular genetic causes of abnormal sexual development, controversies inherent to clinical management and ethical issues.
- Findings in a newborn suggesting the possibility of DSD include (adapted from the American Academy of Pediatrics):
- Apparent male:
- Severe hypospadias associated with bifid scrotum.
- Undescended testis/testes with hypospadias.
- Bilateral non-palpable testes in a full-term apparently male infant.
- Apparent female:
- Clitoral hypertrophy of any degree, non-palpable gonads.
- Vulva with single opening.
- Indeterminate:
- Ambiguous genitalia.
- Any neonate presenting with ambiguous genitalia is an emergency since salt-losing in a 46XX CAH girl can be fatal.
- Apparent male:

